










โพสต์ที่เกี่ยวข้อง

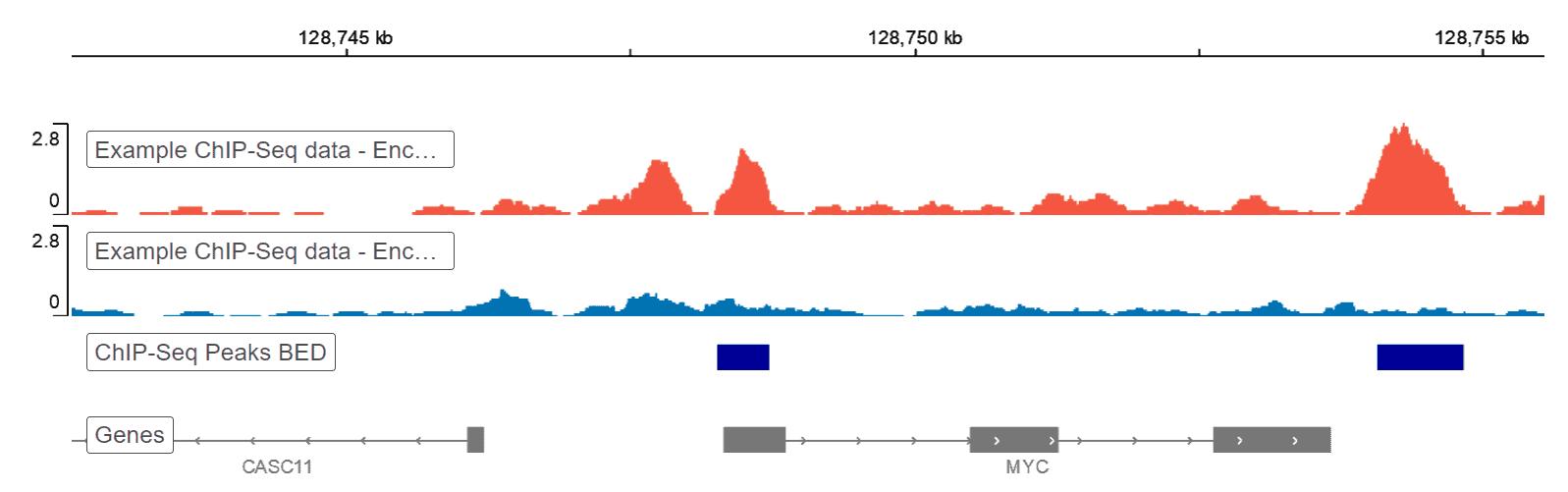

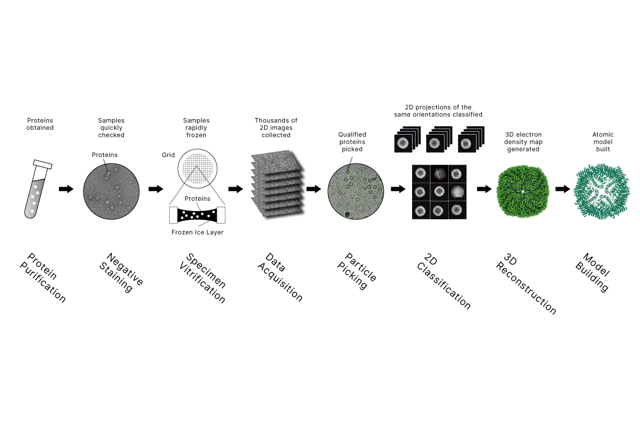
การแก้ไขปัญหา ChIP-Seq 101: แก้ไขสัญญาณต่ําทีละขั้นตอน
2026-02-28การแก้ไขปัญหา ChIP-Seq มักเริ่มต้นด้วยอาการง่ายๆ เพียงอาการเดียว นั่นคือสัญญาณต่ํา แต่สาเหตุที่แท้จริงมักจะกระจายไปตามคุณภาพของโครมาติน ประสิทธิภาพของแอนติบอดี ที่ Longlight Technology เราสนับสนุนห้องปฏิบัติการที่ใช้ ChIP-Seq สําหรับเครื่องหมายฮิสโตนและปัจจัยการถอดความ และเราเห็น "จุดล้มเหลวที่เงียบ" เหมือนกันซ้ําในเครื่องมือและประเภทตัวอย่างที่แตกต่างกัน คู่มือการแก้ไขปัญหา ChIP-Seq 101 นี้จะแนะนําผู้เริ่มต้นผ่านเส้นทางทีละขั้นตอนเพื่อกู้คืนสัญญาณตามลําดับที่สมเหตุสมผล โดยใช้จุดตรวจสอบที่ใช้งานได้จริงที่คุณสามารถตรวจสอบได้ในครั้งเดียว
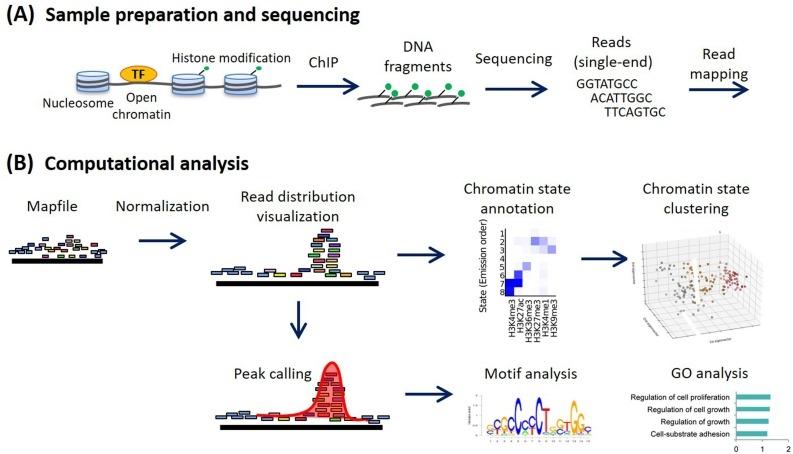
วิธีการวิเคราะห์ ChIP-seq: เวิร์กโฟลว์ที่ใช้งานได้จริงและแอปพลิเคชันขั้นสูง - ScienceDirect
1) กําหนด "สัญญาณต่ํา" ก่อนที่คุณจะเปลี่ยนแปลงอะไร
สัญญาณต่ําอาจหมายถึงสิ่งต่าง ๆ : จุดสูงสุดน้อยเกินไปการเพิ่มคุณค่าที่อ่อนแอที่ตําแหน่งที่รู้จักหรือไลบรารีที่ดูดีแต่แมปไม่ดี การแก้ไขปัญหา ChIP-Seq ที่ดี เริ่มต้นด้วยการติดป้ายกํากับโหมดความล้มเหลวอย่างชัดเจน เนื่องจากแต่ละโหมดชี้ไปที่การแก้ไขที่แตกต่างกัน
วิธีที่เป็นมิตรกับผู้เริ่มต้นในการกําหนดปัญหาคือการตรวจสอบสามชั้น:
✓ ชั้นชีววิทยา: เป้าหมายอยู่ในสถานะเซลล์ของคุณหรือไม่ (กระตุ้นกับการพักผ่อน ระยะความแตกต่าง ระยะเวลาการรักษา) หรือไม่?
✓ ชั้นเสริมคุณค่า: ChIP DNA แสดงการเพิ่มคุณค่าโดย qPCR ที่ตําแหน่งควบคุมเชิงบวกเทียบกับบริเวณลบหรือไม่?
✓เลเยอร์การจัดลําดับ: คุณมีการอ่านแผนที่ที่ไม่ซ้ํากันและระดับที่ซ้ํากันเพียงพอหรือไม่?
หากคุณไม่สามารถตอบสามข้อนี้ได้ เรียกใช้การทดลองที่มีการควบคุมหนึ่งครั้งก่อน: เก็บตัวอย่างไว้เหมือนเดิม ให้ความลึกของการจัดลําดับพอเหมาะ และเปลี่ยนตัวแปรที่น่าสงสัยเพียงตัวเดียว
2) เริ่มต้นด้วยโครมาติน: ขนาดชิ้นส่วน and การควบคุมการสูญเสีย
เมื่อห้องปฏิบัติการถามว่าเหตุใด ChIP จึง "ไม่ทํางาน" สาเหตุที่แท้จริงที่พบบ่อยที่สุดคือโครมาตินที่กระจัดกระจายมากเกินไป ในการแก้ไขปัญหา ChIP-Seq โครมาตินเป็นรากฐานของคุณหากไม่สอดคล้องกันทุกขั้นตอนต่อมาจะกลายเป็นเสียงรบกวน
สําหรับเวิร์กโฟลว์ที่ใช้ sonication ห้องปฏิบัติการหลายแห่งมุ่งเป้าไปที่ชิ้นส่วนในช่วง ~ 150-300 bp สําหรับการเรียกสูงสุดที่คมชัดและการตกตะกอนของภูมิคุ้มกันที่สม่ําเสมอ หากชิ้นส่วนส่วนใหญ่มีขนาดใหญ่กว่า (เช่น >500 bp) แอนติบอดีจะพยายามดึงคอมเพล็กซ์เป้าหมายลงอย่างมีประสิทธิภาพ หากชิ้นส่วนมีขนาดเล็กเกินไป คุณอาจทําลาย epitopes หรือเพิ่มพื้นหลังโดยการปล่อย DNA ที่ไม่จําเพาะ
จุดตรวจที่ใช้งานได้จริงที่คุณสามารถทําได้ทันที:
• วัด DNA หลังจากการเชื่อมโยงย้อนกลับและการล้างข้อมูล (ไม่เพียง แต่ก่อน IP) การลดลงอย่างมากที่นี่ส่งสัญญาณการสูญเสียลูกปัด/คอลัมน์หรือสภาวะที่รุนแรง
•เปรียบเทียบโปรไฟล์การกระจายตัวในตัวอย่าง หากตัวอย่างหนึ่ง "สมบูรณ์แบบ" และตัวอย่างถัดไปมีรอยเปื้อนหรือขนาดใหญ่ ให้เน้นที่การตั้งค่าการสลายและ sonication ไม่ใช่แอนติบอดีก่อน
•เก็บส่วนแบ่ง "อินพุต DNA" จากทุกชุด นี่คือพื้นฐานของคุณสําหรับทั้ง qPCR และ QC ของห้องสมุด
ที่ Longlight Technology เราแนะนําให้ปฏิบัติต่อการเตรียมโครมาตินเหมือนขั้นตอนการผลิตที่มีการควบคุม: แก้ไของค์ประกอบบัฟเฟอร์รักษาอุณหภูมิตัวอย่างให้คงที่ในระหว่างการ sonication และบันทึกการตั้งค่ารอบที่แน่นอน การเบี่ยงเบนเล็กน้อยสร้างความแตกต่างอย่างมากในความแข็งแรงสูงสุดในภายหลัง
3) แอนติบอดีพอดี: ความจําเพาะ, ความแปรผันของล็อต, and การควบคุม
หากโครมาตินดูสมเหตุสมผลขั้นตอนต่อไปในการแก้ไขปัญหา ChIP-Seq คือการเลือกและตรวจสอบแอนติบอดี สัญญาณต่ํามักเกิดจากการใช้แอนติบอดีที่ "ดีสําหรับตะวันตก" แต่อ่อนแอสําหรับ ChIP หรือโดยความแปรปรวนแบบล็อตต่อล็อต
กลยุทธ์แอนติบอดีที่ดีสร้างขึ้นจากการควบคุม:
✓ เป้าหมายการควบคุมเชิงบวก: เครื่องหมายฮิสโตนที่มีการเพิ่มคุณค่าที่แข็งแกร่ง (มักใช้เพื่อยืนยันความสมบูรณ์ของเวิร์กโฟลว์)
✓ การควบคุมเชิงลบ: IgG หรือการควบคุมไอโซไทป์เพื่อประเมินการดึงลงพื้นหลัง
✓ ตําแหน่งที่รู้จัก qPCR: หนึ่งหรือสองตําแหน่งที่เป็นบวกที่เผยแพร่สําหรับเป้าหมายของคุณ รวมถึงพื้นที่ทะเลทรายของยีน
สําหรับปัจจัยการถอดความ สัญญาณอาจต่ํากว่าเครื่องหมายฮิสโตนโดยเนื้อแท้ นั่นหมายความว่าแอนติบอดีของคุณต้องมีความสัมพันธ์สูงและเงื่อนไข IP ของคุณต้องสะอาด หากคุณยังใหม่กับ TF ChIP อย่าเริ่มต้นด้วยการเปลี่ยนความลึกของการจัดลําดับ ขั้นแรกให้ยืนยันการเพิ่มคุณค่าโดย qPCR หากการเพิ่มคุณค่า qPCR อ่อนแอ การอ่านที่มากขึ้นจะทําให้คุณมีสัญญาณรบกวนมากขึ้น
เคล็ดลับที่ใช้ได้จริง: เมื่อคุณเปลี่ยนล็อตแอนติบอดี ให้ตรวจสอบการเพิ่มคุณค่าในชุดโครมาตินเดียวกันอีกครั้งหากเป็นไปได้ หากการเปลี่ยนแปลงล็อตทําให้สัญญาณหยุด เวิร์กโฟลว์ไม่จําเป็นต้อง "ผิด" ประสิทธิภาพของรีเอเจนต์ของคุณเปลี่ยนไป และเส้นทางการแก้ไขปัญหาของคุณควรสะท้อนถึงสิ่งนั้น
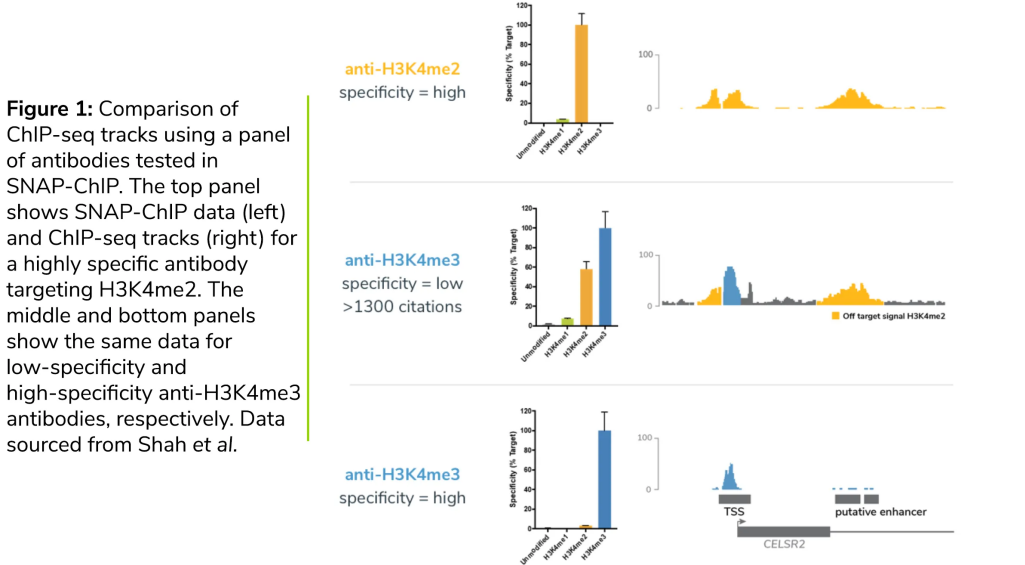
การเลือกแอนติบอดี ChIP ที่เหมาะสมสําหรับการทดลองของคุณ - EpiCypher
4) ลําดับการเพิ่มประสิทธิภาพ IP ที่สะอาด—ไม่ต้องคาดเดา
นอกเหนือจากโครมาตินและแอนติบอดีแล้ว เคมี IP (ลูกปัด การล้าง การฟักไข่) เป็นฮอตสปอตต่อไป สัญญาณต่ํามักเป็นปัญหาเบื้องหลัง
✓ การเลือกลูกปัด: เลือกโปรตีน A/G ที่เหมาะกับสายพันธุ์แอนติบอดี/ไอโซไทป์ของคุณ
✓ ปริมาณแอนติบอดี: ไทเทรตเพื่อหลีกเลี่ยงการดึงลงที่อ่อนแอหรือดีเอ็นเอที่ไม่จําเพาะสูงขึ้น
✓ สมดุลการซัก: ปรับเทียบความเข้มงวดเพื่อขจัดเสียงรบกวน แต่ยังคงปฏิสัมพันธ์ที่อ่อนแอแต่เป็นจริง
กฎสําหรับผู้เริ่มต้นที่ใช้งานได้จริงคือการปรับเพียงแกนเดียวต่อการวิ่ง:
•หากมีจุดสูงสุดอยู่ แต่อ่อนแอให้เพิ่มการจับที่มีประสิทธิภาพ (แอนติบอดีมากขึ้นเล็กน้อยการผูกลูกปัดที่ดีขึ้นการฟักตัวนานขึ้น)
•หากจุดสูงสุดกว้างและมีเสียงดังให้เพิ่มความจําเพาะ (การล้างที่แรงขึ้นการปิดกั้นที่ดีขึ้นลดแอนติบอดีมากเกินไป)
จากมุมมองของผู้ผลิต Longlight Technology ออกแบบรีเอเจนต์การตกตะกอนของภูมิคุ้มกันและระบบลูกปัดแม่เหล็กเพื่อลดการสูญเสียระหว่างการจัดการ เนื่องจากการสูญเสียตัวอย่างมีลักษณะเหมือน "สัญญาณต่ํา" ทุกประการ การแยกลูกปัดที่ราบรื่น การผูกมัดที่สม่ําเสมอ และขั้นตอนการล้างที่สะอาดช่วยลดความแปรปรวนระหว่างผู้ปฏิบัติงาน ซึ่งเป็นสิ่งสําคัญอย่างยิ่งสําหรับทีมที่ฝึกอบรมพนักงานใหม่
5) ห้องสมุด QC: เมื่อ "ดีเอ็นเอที่ดี" ยังคงให้สัญญาณต่ํา
บางครั้งการเพิ่มคุณค่า DNA ของ ChIP ก็มีจริง แต่ข้อมูลสุดท้ายยังคงดูแบนราบ ในการแก้ไขปัญหา ChIP-Seq สิ่งนี้มักจะชี้ไปที่การสร้างไลบรารีหรือเมตริกการจัดลําดับ
สาเหตุทั่วไปในระดับไลบรารีของสัญญาณต่ํา:
✓ Over-amplification: รอบ PCR มากเกินไปสามารถขยายรายการที่ซ้ํากันและลดการอ่านที่ไม่ซ้ํากันที่ใช้งานได้
✓สิ่งประดิษฐ์อะแดปเตอร์ / ไพรเมอร์: สิ่งเหล่านี้ใช้การอ่านลําดับโดยไม่ปรับปรุงความครอบคลุมเป้าหมาย
✓ ความซับซ้อนไม่ดี: มักเกิดจากการป้อนข้อมูล ChIP DNA ต่ํามากหรือการสูญหายระหว่างการล้างข้อมูล
สิ่งที่ต้องตรวจสอบก่อนที่คุณจะเรียกใช้ ChIP ทั้งหมดอีกครั้ง:
•การกระจายขนาดห้องสมุด (คุณต้องการจุดสูงสุดที่สะอาดไม่ใช่จุดสูงสุดที่ไม่คาดคิดหลายจุด)
•อัตราที่ซ้ํากันและอัตราการทําแผนที่ที่ไม่ซ้ํากันหลังจากการจัดตําแหน่ง
•เศษส่วนของการอ่านในแนวโน้มสูงสุด (FRiP) ที่สัมพันธ์กับพื้นฐานภายในของคุณ (แม้แต่ผู้เริ่มต้นก็สามารถติดตาม "ดีขึ้นและแย่ลง" ตลอดการวิ่ง)
หากคุณสงสัยว่าไลบรารีมีการหมุนเวียนมากเกินไปการปรับปรุงง่ายๆคือการลดจํานวนรอบและเพิ่มประสิทธิภาพการจับภาพต้นน้ํา (การกู้คืนโครมาตินที่ดีขึ้นและความจําเพาะของ IP) การจัดลําดับที่ลึกกว่านี้ไม่สามารถชดเชยไลบรารีที่มีความซับซ้อนต่ําได้
6) ขั้นตอน-โดย-การกู้คืนสัญญาณขั้นตอนที่คุณสามารถเริ่มได้ในวันพรุ่งนี้
ลําดับที่ใช้งานได้จริงที่มีประสิทธิภาพเหนือกว่าการปรับแต่งเฉพาะกิจ:
✓ ขั้นตอนที่ 1: ตรวจสอบว่าชิ้นส่วนโครมาตินมี ~150–300 bp และยืนยันการกู้คืน DNA หลังการเชื่อมโยงย้อนกลับ
✓ ขั้นตอนที่ 2: ตรวจสอบการเพิ่มคุณค่าด้วย qPCR ที่ตําแหน่งบวกหนึ่งตําแหน่งและลบหนึ่งตําแหน่งก่อนการเตรียมห้องสมุด
✓ ขั้นตอนที่ 3: เพิ่มการควบคุมที่เหมาะสม (อินพุต IgG) เพื่อแยก "ไม่มีการเพิ่มคุณค่า" ออกจาก "พื้นหลังสูง"
✓ ขั้นตอนที่ 4: ปรับเงื่อนไข IP ทีละตัวแปร (ลูกปัด ปริมาณแอนติบอดี ความเข้มงวดในการซัก)
✓ ขั้นตอนที่ 5: ตรวจสอบตัวชี้วัดไลบรารี (การทําสําเนา การทําแผนที่ การกระจายขนาด) ก่อนที่จะสันนิษฐานว่าความลึกของการจัดลําดับเป็นปัญหา
CTA (เทคโนโลยี Longlight): หากคุณต้องการเส้นทางที่เร็วขึ้น โปรดติดต่อ Longlight Technology เพื่อขอรายการตรวจสอบการแก้ไขปัญหา ChIP-Seq และแผ่นงานการวินิจฉัยแบบตัวอย่างต่อตัวอย่าง (ไลบรารีโครมาติน→ IP →) นอกจากนี้เรายังสามารถแนะนําการออกแบบการควบคุมและกลยุทธ์การจับคู่รีเอเจนต์เพื่อลดความแปรปรวนของผู้ปฏิบัติงานและช่วยให้ผู้เริ่มต้นบรรลุการเพิ่มคุณค่าที่เสถียรได้เร็วขึ้น
สัญญาณต่ําน่าหงุดหงิด แต่ไม่ค่อยลึกลับ ด้วยขั้นตอนการแก้ไขปัญหา ChIP-Seq ที่มีระเบียบวินัย โดยเริ่มจากความสมบูรณ์ของโครมาติน จากนั้นจึงปรับแอนติบอดี ตามด้วยความจําเพาะของ IP และสุดท้ายคือ QC ของไลบรารี คุณสามารถเปลี่ยนการเรียกใช้ที่อ่อนแอเพียงครั้งเดียวให้เป็นโปรโตคอลที่ทําซ้ําได้ซึ่งปรับขนาดตามตัวอย่าง พนักงาน และโครงการ